
📖 Step 2 — Learning Material
🔹 1️⃣ Introduction
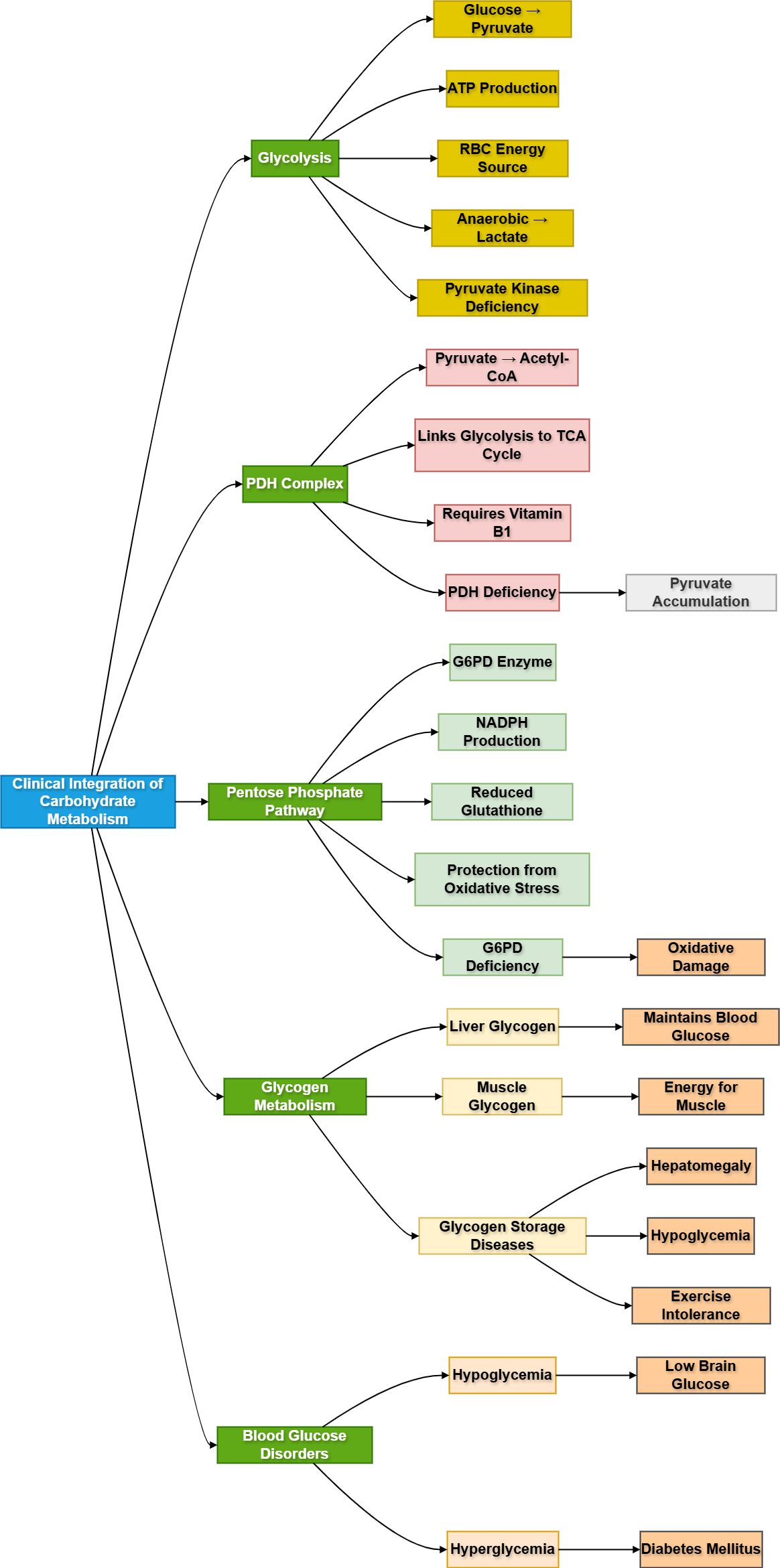
Carbohydrate metabolism is central to human energy production and blood glucose regulation. Glucose is used by almost every tissue, but it is especially important for the brain, red blood cells, liver, and skeletal muscle. In 2nd Year MBBS, students must understand not only the pathways but also how failure of these pathways produces disease. Glycolysis, PDH activity, G6PD function, glycogen metabolism, hypoglycemia, and hyperglycemia are clinically important because they explain common presentations such as fatigue, seizures, jaundice, hemolysis, lactic acidosis, and diabetes mellitus. This topic integrates basic biochemistry with clinical medicine so students can understand “why the patient develops symptoms.”
🔹 2️⃣ Foundation Concepts
Key Definitions
- Carbohydrate metabolism: Biochemical processing of glucose and related sugars for energy production, storage, and blood glucose maintenance.
- Glycolysis: Cytoplasmic pathway that converts glucose into pyruvate with ATP production.
- Aerobic glycolysis: Glycolysis followed by pyruvate entry into mitochondria for aerobic metabolism.
- Anaerobic glycolysis: Conversion of pyruvate into lactate when oxygen or mitochondria are unavailable.
- PDH complex: Mitochondrial enzyme complex that converts pyruvate into acetyl-CoA.
- G6PD: Enzyme of the pentose phosphate pathway that produces NADPH.
- NADPH: Reducing power required to protect red blood cells from oxidative damage.
- Glycogen: Storage form of glucose, mainly in liver and skeletal muscle.
- Hypoglycemia: Abnormally low blood glucose.
- Hyperglycemia: Abnormally high blood glucose.
🔹 3️⃣ Core Learning — Curriculum Coverage
1: Clinical Correlations of Glycolysis
🧠 CORE
- Glycolysis occurs in the cytoplasm of all cells.
- It converts glucose into pyruvate.
- Net energy yield is 2 ATP and 2 NADH per glucose.
- It is the only ATP source in red blood cells.
- Under anaerobic conditions, pyruvate converts into lactate.
- Important glycolytic enzymes include hexokinase/glucokinase, PFK-1, and pyruvate kinase.
- Defects in glycolysis may cause fatigue, muscle weakness, hemolysis, and lactic acidosis.
- Cancer cells commonly show increased glycolysis even in oxygen presence, called the Warburg effect.
🔬 CONCEPT EXPLAINED
Glycolysis exists to provide quick ATP from glucose. It is especially useful in tissues that either lack mitochondria, like red blood cells, or have high energy demand, like exercising skeletal muscle.
In red blood cells, glycolysis is essential because RBCs have no mitochondria. Therefore, they cannot use the TCA cycle or oxidative phosphorylation. If glycolysis fails, RBC membrane ion pumps fail due to ATP deficiency, causing membrane rigidity and hemolysis.
In skeletal muscle during intense exercise, oxygen delivery may be insufficient. Pyruvate is then converted into lactate to regenerate NAD⁺, allowing glycolysis to continue.
⚠️ What Happens If It Fails?
- Pyruvate kinase deficiency → reduced ATP in RBCs → chronic hemolytic anemia.
- Excess anaerobic glycolysis → lactate accumulation → lactic acidosis.
- Poor glucose utilization → fatigue and weakness.
🩺 Clinical Link
A child with chronic hemolytic anemia, jaundice, and splenomegaly may have pyruvate kinase deficiency because RBCs depend entirely on glycolysis for ATP.
2: PDH Deficiency
🧠 CORE
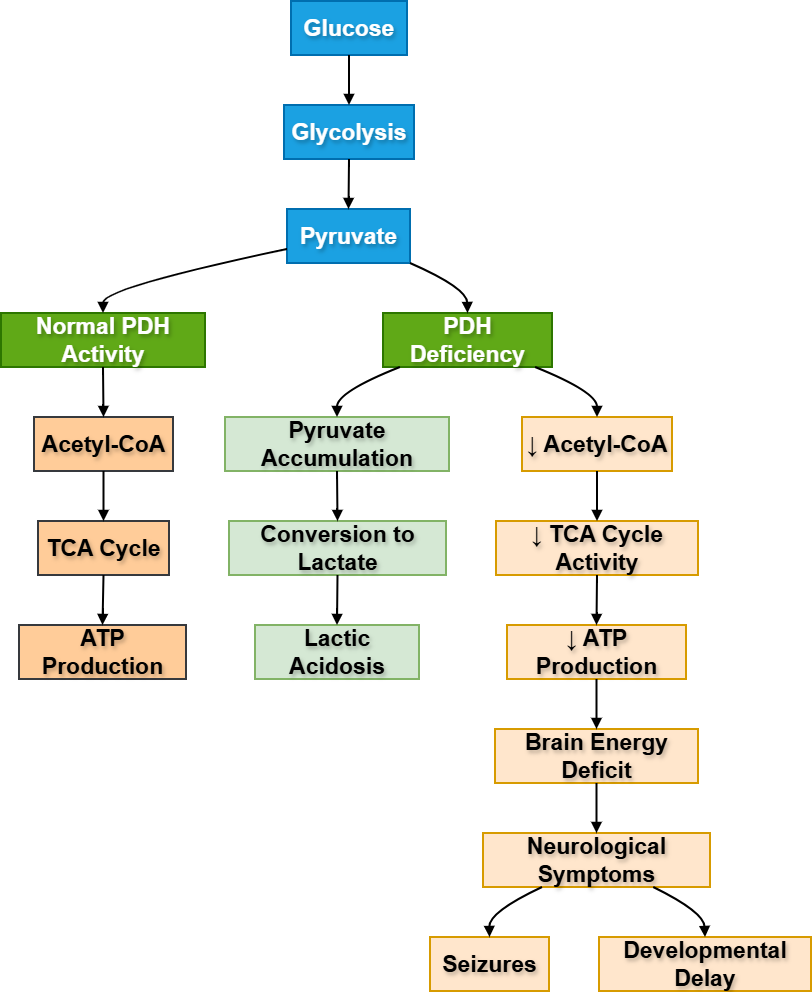
- PDH means pyruvate dehydrogenase complex.
- It is located in the mitochondria.
- It converts pyruvate into acetyl-CoA.
- Acetyl-CoA enters the TCA cycle for energy production.
- PDH links glycolysis with aerobic metabolism.
- PDH requires vitamins: B1, B2, B3, B5, and lipoic acid.
- PDH deficiency causes pyruvate accumulation.
- Excess pyruvate is converted into lactate.
- Main clinical result is lactic acidosis with neurological dysfunction.
🔬 CONCEPT EXPLAINED
PDH acts like a metabolic bridge. Glycolysis produces pyruvate in the cytoplasm. For complete aerobic oxidation, pyruvate must enter mitochondria and become acetyl-CoA. PDH performs this conversion.
If PDH is deficient, pyruvate cannot efficiently enter the TCA cycle. As a result, pyruvate accumulates and is converted into lactate. This causes lactic acidosis. Since the brain depends heavily on aerobic glucose metabolism, neurological symptoms are common.
⚠️ What Happens If It Fails?
- Pyruvate cannot become acetyl-CoA.
- TCA cycle activity decreases.
- ATP production decreases.
- Lactate increases.
- Patient develops lactic acidosis, developmental delay, seizures, and neurological problems.
🩺 Clinical Link
PDH deficiency should be suspected in an infant with neurological symptoms and persistent lactic acidosis, especially when blood glucose may be normal but energy production is impaired.
3: G6PD Deficiency
🧠 CORE
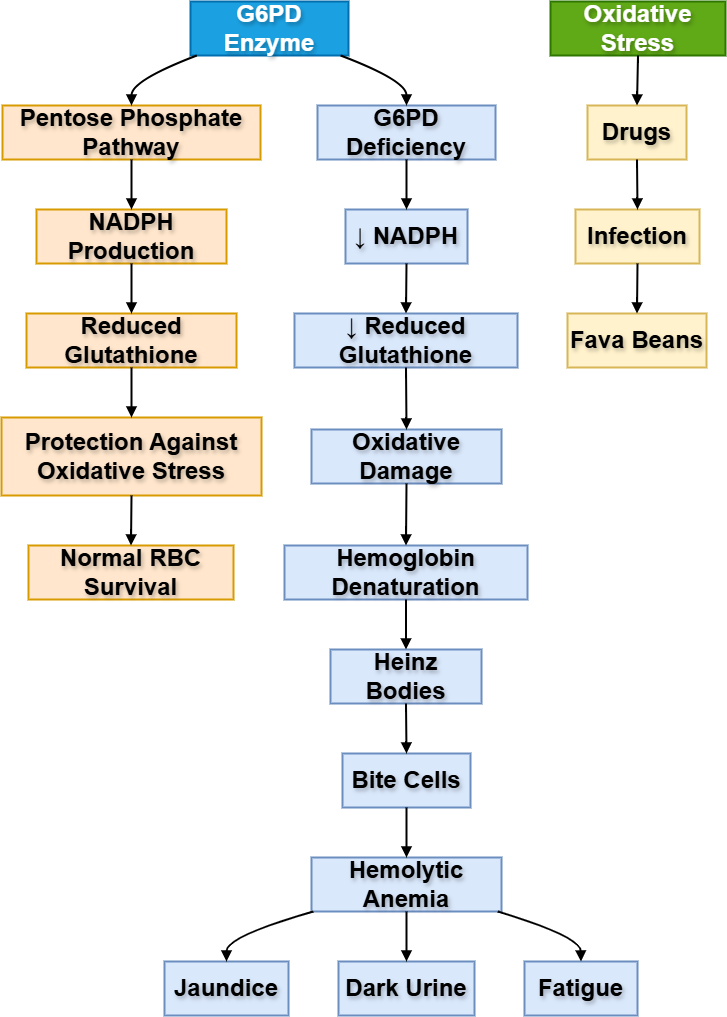
- G6PD is an enzyme of the pentose phosphate pathway.
- It produces NADPH.
- NADPH keeps glutathione in reduced form.
- Reduced glutathione protects RBCs from oxidative stress.
- RBCs are highly vulnerable because they lack mitochondria.
- G6PD deficiency is commonly X-linked.
- Oxidative triggers include infections, certain drugs, and fava beans.
- It causes episodic hemolytic anemia.
- Blood smear may show Heinz bodies and bite cells.
🔬 CONCEPT EXPLAINED
The main purpose of G6PD in RBCs is protection, not ATP production. RBCs continuously face oxidative stress because they carry oxygen. NADPH produced by the pentose phosphate pathway maintains reduced glutathione, which neutralizes oxidants.
If G6PD is deficient, RBCs cannot handle oxidative stress. Hemoglobin becomes damaged and forms Heinz bodies. Splenic macrophages remove parts of damaged RBCs, producing bite cells. Eventually, RBCs undergo hemolysis.
⚠️ What Happens If It Fails?
- NADPH decreases.
- Reduced glutathione decreases.
- Oxidative damage increases.
- Hemoglobin denatures.
- RBCs rupture.
- Patient develops jaundice, dark urine, pallor, fatigue, and hemolytic anemia.
🩺 Clinical Link
A young male who develops jaundice and dark urine after taking an oxidant drug or after infection may have G6PD deficiency.
4: Glycogen Storage Diseases
🧠 CORE
- Glycogen is the storage form of glucose.
- Liver glycogen maintains blood glucose during fasting.
- Muscle glycogen provides energy during muscle contraction.
- Glycogen storage diseases (GSDs) occur due to inherited enzyme defects in glycogen metabolism.
- Defective glycogen breakdown or synthesis causes abnormal glycogen accumulation.
- Liver GSDs commonly produce fasting hypoglycemia and hepatomegaly.
- Muscle GSDs commonly produce exercise intolerance and muscle cramps.
- Some GSDs affect lysosomes causing generalized tissue involvement.
- Important GSDs include:
- Von Gierke disease (Type I)
- Pompe disease (Type II)
- Cori disease (Type III)
- McArdle disease (Type V)
🔬 CONCEPT EXPLAINED
Glycogen serves as a rapidly mobilizable glucose reserve. After meals, excess glucose is converted into glycogen mainly in liver and skeletal muscle.
Structure → Function
- Liver glycogen
- Maintains blood glucose during fasting.
- Contains glucose-6-phosphatase allowing free glucose release into blood.
- Muscle glycogen
- Provides ATP for muscle contraction during exercise.
- Lacks glucose-6-phosphatase, so glucose remains for local muscle use.
If enzymes involved in glycogen degradation are defective, glycogen accumulates abnormally inside cells and glucose release becomes impaired.
Important Glycogen Storage Diseases
1. Von Gierke Disease (Type I)
Enzyme Defect:
Glucose-6-phosphatase deficiency
Main Organ:
Liver
Mechanism:
- Liver cannot convert glucose-6-phosphate into free glucose.
- Glycogen breakdown occurs partially, but glucose cannot enter blood.
- Severe fasting hypoglycemia develops.
Key Features:
- Hepatomegaly
- Severe fasting hypoglycemia
- Lactic acidosis
- Hyperuricemia
- Hyperlipidemia
Why Hepatomegaly Occurs:
Excess glycogen accumulates inside hepatocytes.
2. Pompe Disease (Type II)
Enzyme Defect:
Lysosomal acid α-1,4-glucosidase (acid maltase) deficiency
Main Organ:
Heart and muscle
Mechanism:
- Glycogen accumulates inside lysosomes.
- Lysosomal enlargement damages muscle cells.
Key Features:
- Cardiomegaly
- Muscle weakness
- Hypotonia
- Heart failure in infants
Unique Point:
This is a lysosomal storage disease.
3. Cori Disease (Type III)
Enzyme Defect:
Debranching enzyme deficiency
Main Organ:
Liver and muscle
Mechanism:
- Glycogen breakdown remains incomplete.
- Abnormal branched glycogen accumulates.
Key Features:
- Mild hypoglycemia
- Hepatomegaly
- Muscle weakness
Important Difference from Von Gierke:
Hypoglycemia is less severe.
4. McArdle Disease (Type V)
Enzyme Defect:
Muscle glycogen phosphorylase deficiency
Main Organ:
Skeletal muscle
Mechanism:
- Muscle cannot utilize glycogen during exercise.
- ATP production during exertion becomes inadequate.
Key Features:
- Exercise intolerance
- Muscle cramps
- Myoglobinuria after exercise
Important Point:
Blood glucose usually remains normal because liver is unaffected.
5. Hers Disease (Glycogen Storage Disease Type VI)
Hers disease is a glycogen storage disease caused by deficiency of liver glycogen phosphorylase, the enzyme responsible for glycogen breakdown in the liver. Because glycogen cannot be properly converted into glucose, affected children develop mild fasting hypoglycemia and hepatomegaly (enlarged liver) due to glycogen accumulation.
Key Features
- Autosomal recessive disorder
- Defect in liver glycogen phosphorylase
- Glycogen accumulates mainly in liver
- Mild fasting hypoglycemia
- Hepatomegaly
- Growth retardation may occur
- Usually milder than Von Gierke disease
Important Concept
Muscle glycogen breakdown remains normal, so muscle symptoms are usually absent.
Diagnosis
- Elevated liver glycogen
- Mild ketosis during fasting
- Enzyme assay or genetic testing
Treatment
- Frequent carbohydrate-rich meals
- Avoid prolonged fasting
- High-protein diet may help maintain blood glucose levels
⚠️ What Happens If It Fails?
Liver Enzyme Defect
↓ Glucose release into blood
→ Fasting hypoglycemia
→ Glycogen accumulation
→ Hepatomegaly
Muscle Enzyme Defect
↓ ATP production during exercise
→ Muscle cramps
→ Exercise intolerance
Lysosomal Enzyme Defect
↓ Glycogen degradation in lysosomes
→ Cellular enlargement and dysfunction
→ Cardiomyopathy and muscle weakness
🩺 Clinical Link
Von Gierke Disease
Child with:
- Severe fasting hypoglycemia
- Enlarged liver
- Lactic acidosis
Suggests glucose-6-phosphatase deficiency.
Pompe Disease
Infant with:
- Cardiomegaly
- Hypotonia
- Heart failure
Suggests lysosomal glycogen accumulation.
McArdle Disease
Young adult with:
- Muscle cramps after exercise
- Myoglobinuria
- Poor exercise tolerance
Suggests muscle glycogen phosphorylase deficiency.
5: Metabolic Causes of Hypoglycemia
🧠 CORE
- Hypoglycemia means low blood glucose.
- Brain depends mainly on glucose.
- Symptoms occur due to neuroglycopenia and sympathetic activation.
- Important causes include prolonged fasting, excess insulin, liver disease, alcohol intake, adrenal insufficiency, and glycogen storage disease.
- Newborns and children are more vulnerable.
- Liver is the main organ that prevents fasting hypoglycemia.
- Glucagon and epinephrine increase blood glucose.
- Severe hypoglycemia may cause seizures, coma, or death.
🔬 CONCEPT EXPLAINED
Blood glucose is maintained by glycogenolysis and gluconeogenesis. During early fasting, liver glycogen breaks down to release glucose. During prolonged fasting, gluconeogenesis becomes more important.
Hypoglycemia occurs when glucose utilization is increased, glucose production is reduced, or insulin action is excessive. Since the brain cannot store significant glucose, neurological symptoms appear quickly.
⚠️ What Happens If It Fails?
- Brain ATP decreases.
- Patient develops confusion, irritability, sweating, tremors, seizures, or coma.
- Severe prolonged hypoglycemia can cause permanent brain injury.
🩺 Clinical Link
A diabetic patient taking insulin who misses a meal may develop sweating, tremors, confusion, and unconsciousness due to hypoglycemia.
6: Metabolic Causes of Hyperglycemia
🧠 CORE
- Hyperglycemia means high blood glucose.
- Most important cause is diabetes mellitus.
- It results from insulin deficiency, insulin resistance, or both.
- Insulin promotes glucose uptake in muscle and adipose tissue through GLUT-4.
- Insulin also promotes glycogenesis and inhibits gluconeogenesis.
- In diabetes, glucose remains in blood instead of entering cells properly.
- Hyperglycemia causes osmotic diuresis.
- Chronic hyperglycemia damages blood vessels and nerves.
- Clinical features include polyuria, polydipsia, weight loss, fatigue, and recurrent infections.
🔬 CONCEPT EXPLAINED
Insulin is the main hormone of the fed state. It tells the body to use and store glucose. When insulin is absent or ineffective, glucose cannot enter insulin-dependent tissues efficiently. The liver also continues producing glucose by gluconeogenesis and glycogenolysis.
As blood glucose rises above renal threshold, glucose appears in urine. Glucose pulls water with it, causing polyuria. Water loss causes polydipsia. Cells behave as if they are starving despite high blood glucose, causing weight loss and fatigue.
⚠️ What Happens If It Fails?
- Glucose uptake decreases.
- Hepatic glucose production increases.
- Blood glucose rises.
- Glucosuria and dehydration occur.
- Long-term complications include retinopathy, nephropathy, neuropathy, and vascular disease.
🩺 Clinical Link
A patient with polyuria, polydipsia, weight loss, and high random blood glucose most likely has diabetes mellitus due to disturbed carbohydrate metabolism.
⚙️ 4️⃣ Functional Flow
Carbohydrate metabolism is not a set of isolated pathways. It works as an integrated system.
- Glycolysis provides immediate ATP.
- PDH connects glycolysis to mitochondrial energy production.
- Pentose phosphate pathway protects RBCs through NADPH.
- Glycogen metabolism maintains blood glucose during fasting and supports muscle activity.
- Insulin and glucagon regulate blood glucose according to fed and fasting states.
- Failure of these systems produces recognizable clinical syndromes such as hemolysis, lactic acidosis, hypoglycemia, and hyperglycemia.
-
Common Exam Traps
- Do not confuse G6PD deficiency with pyruvate kinase deficiency.
- G6PD → ↓ NADPH → oxidative hemolysis
- Pyruvate kinase → ↓ ATP → membrane instability
- Do not say muscle glycogen maintains blood glucose.
- Muscle lacks glucose-6-phosphatase.
- PDH deficiency does not directly cause hypoglycemia.
- It mainly causes lactic acidosis and neurological dysfunction.
- Hyperglycemia in diabetes is due to both:
- decreased peripheral glucose uptake
- increased hepatic glucose production
- Lactate rises when pyruvate cannot enter aerobic metabolism properly.
📌 6️⃣ Summary Points
- RBCs depend completely on glycolysis for ATP.
- PDH deficiency causes lactic acidosis because pyruvate converts into lactate.
- G6PD deficiency causes hemolysis due to oxidative stress.
- Liver glycogen maintains blood glucose; muscle glycogen is for muscle use only.
- Hypoglycemia mainly affects the brain.
- Hyperglycemia mainly reflects insulin deficiency or insulin resistance.
- Diabetes is the most important clinical example of disturbed carbohydrate metabolism.
🎥 Recommended Integrated Video Playlist
Clinical Integration of Carbohydrate Metabolism — Glycolysis, PDH Deficiency, G6PD Deficiency, Glycogen Storage Diseases, Hypoglycemia & Hyperglycemia