
📖 Step 2 — Learning Material
🔹 1️⃣ Introduction
Proteins are continuously synthesized and broken down in the body, producing amino acids that enter a common metabolic reservoir called the amino acid pool. This pool is essential for growth, tissue repair, energy production, and synthesis of important biomolecules. Excess amino acids cannot be stored in the body, therefore their nitrogen must be safely removed through protein catabolism and converted into urea in the liver.
Nitrogen metabolism mainly occurs in the liver, while skeletal muscle plays a major role in amino acid breakdown and ammonia transport. The urea cycle protects the body from ammonia toxicity, especially toxicity affecting the brain.
Clinically, disturbances in amino acid metabolism or defects in the urea cycle can cause hyperammonemia, neurological dysfunction, liver disease manifestations, and inherited metabolic disorders. Understanding this topic forms the biochemical basis for liver function tests, transaminase interpretation, hepatic encephalopathy, and inborn errors of metabolism.
🔹 2️⃣ Foundation Concepts
Key Definitions
- Amino Acid Pool: Total free amino acids present in body fluids and cells.
- Protein Catabolism: Breakdown of proteins into amino acids and nitrogenous waste.
- Transamination: Transfer of amino group from one amino acid to another keto acid.
- Deamination: Removal of amino group as free ammonia.
- Transdeamination: Combination of transamination and oxidative deamination.
- Ammonia (NH₃): Toxic nitrogen-containing compound formed during amino acid breakdown.
- Urea Cycle: Metabolic pathway converting toxic ammonia into urea in liver.
- Transaminases: Enzymes catalyzing transamination reactions (ALT and AST).
Essential Terminology
- Aminotransferase
- Glutamate
- α-ketoglutarate
- Oxidative deamination
- Hyperammonemia
- Carbamoyl phosphate
- Ornithine cycle
- Hepatic encephalopathy
Basic Overview
- Body proteins are constantly degraded and synthesized.
- Amino acids enter a common amino acid pool.
- Excess amino acids are catabolized because they cannot be stored.
- Amino group removal produces ammonia.
- Ammonia is highly toxic, especially to brain tissue.
- Liver converts ammonia into urea through urea cycle.
- Urea is excreted by kidneys.
🔹 3️⃣ Core Learning — Curriculum Coverage
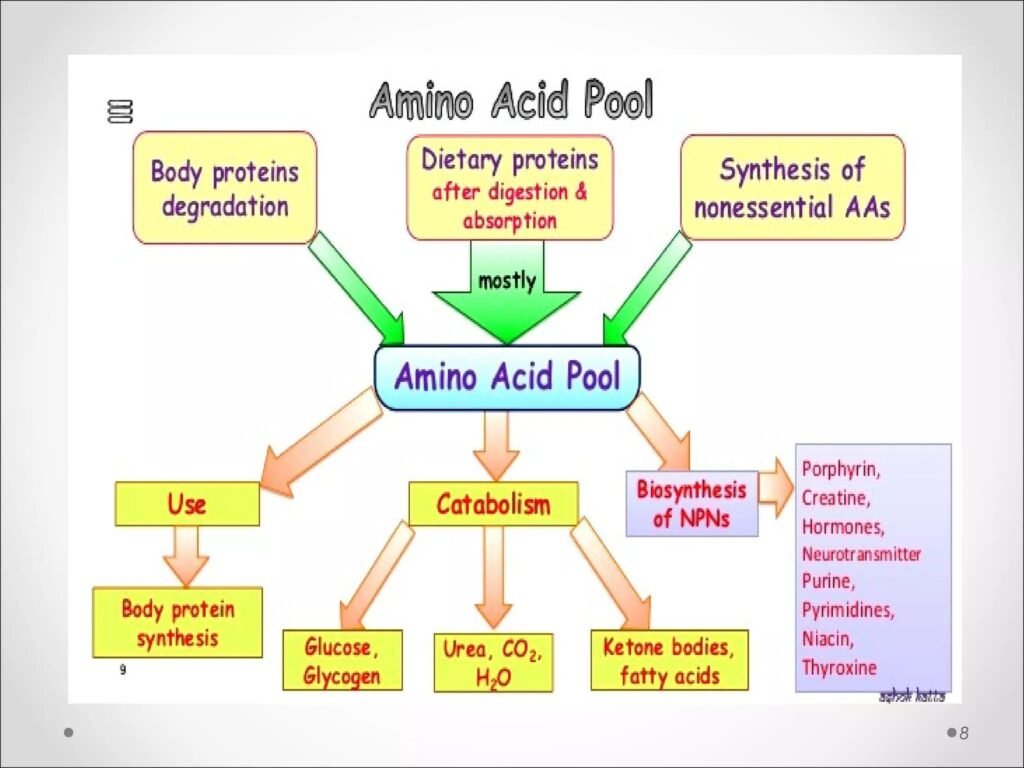
A. Amino Acid Pool
🧠 CORE
Definition
The amino acid pool is the total quantity of free amino acids available in body tissues and circulation for metabolic use.
Major Components
- Free amino acids in plasma
- Intracellular amino acids
- Dietary amino acids
- Amino acids from tissue protein breakdown
- Nonessential amino acids synthesized in body
Primary Function
- Protein synthesis
- Formation of specialized compounds
- Energy production
- Nitrogen transport
- Glucose and fat synthesis during starvation
Sources of Amino Acids Entering Pool
- Dietary protein digestion
- Breakdown of body proteins
- Synthesis of nonessential amino acids
Utilization of Amino Acids from Pool
- Synthesis of tissue proteins
- Formation of hormones and neurotransmitters
- Energy production
- Gluconeogenesis
- Lipogenesis
- Nitrogen-containing compound synthesis
🔬 CONCEPT EXPLAINED
The body has no storage form for excess amino acids similar to glycogen or fat. Therefore, all amino acids circulate in a dynamic metabolic reservoir called the amino acid pool.
After digestion, proteins are broken into amino acids and absorbed from the intestine into blood. Tissue proteins also undergo continuous turnover, releasing amino acids back into circulation.
Cells withdraw amino acids from the pool whenever needed for:
- New protein synthesis
- Enzyme formation
- Hormone synthesis
- Tissue repair
- Energy generation
When amino acid intake exceeds requirement, excess amino acids are degraded. Their carbon skeleton enters energy pathways while nitrogen is converted into urea.
Structure → Function Relationship
- Continuous protein turnover maintains pool stability.
- Liver acts as major regulator of amino acid metabolism.
- Muscle contributes large amounts of amino acids during fasting.
⚠️ IF DAMAGED
Cause → Effect
- Severe liver disease → impaired amino acid metabolism → hyperammonemia
- Protein malnutrition → reduced amino acid pool → muscle wasting
- Excessive protein breakdown → increased ammonia production
- Starvation → muscle protein breakdown → negative nitrogen balance
B. Protein Catabolism Processes
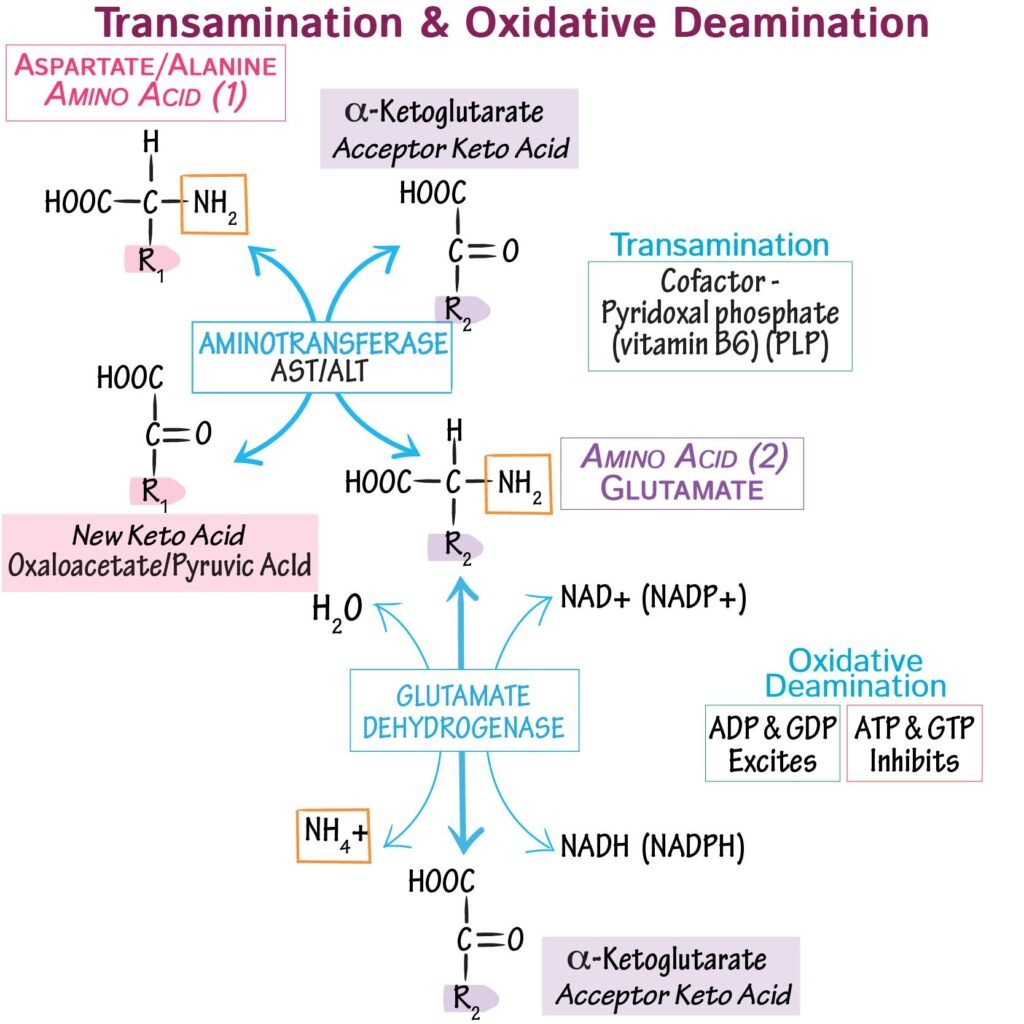
1. Transamination
🧠 CORE
Definition
Transfer of amino group from an amino acid to a keto acid.
Major Components
- Aminotransferase enzymes
- Pyridoxal phosphate (Vitamin B₆)
- Amino acid donor
- α-keto acid acceptor
Primary Function
- Collection of amino groups
- Formation of glutamate
- Synthesis of nonessential amino acids
Important Enzymes
- ALT (Alanine transaminase)
- AST (Aspartate transaminase)
🔬 CONCEPT EXPLAINED
Most amino acids first undergo transamination before nitrogen removal.
In this reaction:
- Amino group is transferred to α-ketoglutarate
- Glutamate is formed
- Original amino acid becomes corresponding keto acid
Example:
Alanine + α-ketoglutarate → Pyruvate + Glutamate
Vitamin B₆ acts as coenzyme for all transaminases.
Why It Exists
- Safely channels amino groups toward glutamate
- Prevents direct release of toxic ammonia
- Allows amino acid interconversion
Structure → Function
- Liver cells contain high transaminase activity.
- Cytoplasm and mitochondria support rapid amino acid metabolism.
⚠️ IF DAMAGED
- Vitamin B₆ deficiency → impaired transamination
- Liver cell injury → ALT and AST leak into blood
- Severe hepatic damage → impaired amino acid metabolism
2. Deamination
🧠 CORE
Definition
Removal of amino group from amino acid as free ammonia.
Major Components
- Glutamate dehydrogenase
- NAD⁺ or NADP⁺
- Glutamate
Primary Function
- Release ammonia for urea synthesis
- Regenerate α-ketoglutarate
🔬 CONCEPT EXPLAINED
Oxidative deamination mainly occurs in liver mitochondria.
Glutamate loses amino group and produces:
- Free ammonia
- α-ketoglutarate
Reaction:
Glutamate → α-ketoglutarate + NH₃
Why It Exists
- Removes excess nitrogen
- Provides ammonia for urea cycle
Structure → Function
- Liver mitochondria contain glutamate dehydrogenase.
- Mitochondrial location helps rapid ammonia detoxification.
⚠️ IF DAMAGED
- Liver failure → ammonia accumulation
- Excess ammonia → brain toxicity
- Reduced deamination → impaired nitrogen disposal
3. Transdeamination
🧠 CORE
Definition
Combined process of transamination followed by oxidative deamination.
Primary Function
- Major pathway for amino acid catabolism
- Efficient nitrogen removal
🔬 CONCEPT EXPLAINED
Most amino acids:
- Transfer amino group to α-ketoglutarate
- Form glutamate
- Glutamate undergoes oxidative deamination
- Ammonia enters urea cycle
Thus:
- Nitrogen is safely collected
- Ammonia production becomes controlled
⚠️ IF DAMAGED
- Defective liver metabolism → ammonia accumulation
- Impaired urea synthesis → hyperammonemia
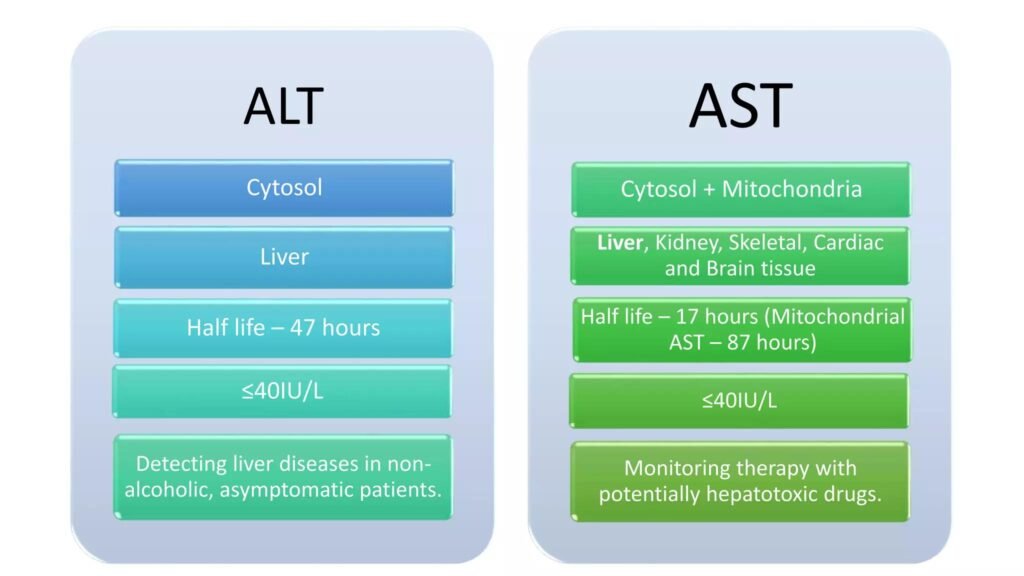
4. Clinical Importance of Transaminases
🧠 CORE
Important Enzymes
- ALT (SGPT)
- AST (SGOT)
Clinical Use
- Indicators of liver cell injury
- Used in liver function assessment
🔬 CONCEPT EXPLAINED
Transaminases are intracellular enzymes.
When hepatocytes are damaged:
- Cell membranes rupture
- ALT and AST leak into blood
ALT is more liver-specific.
AST is also found in:
- Heart
- Skeletal muscle
- Liver
Why It Exists
These enzymes normally participate in amino acid metabolism but clinically act as biomarkers of tissue damage.
⚠️ IF DAMAGED
- Viral hepatitis → markedly raised ALT/AST
- Myocardial infarction → elevated AST
- Severe liver necrosis → very high enzyme levels
C. Ammonia Formation and Transport
🧠 CORE
Formation of Ammonia
- Amino acid catabolism
- Intestinal bacterial action
- Purine and pyrimidine metabolism
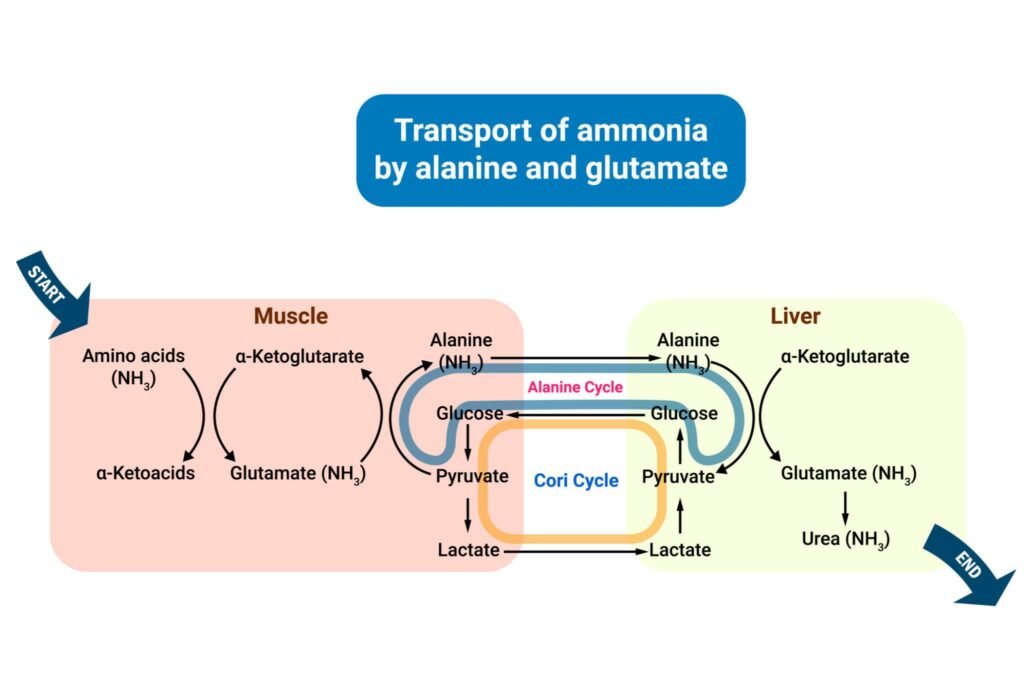
Transport Forms
- Glutamine
- Alanine
Primary Function
- Safe transport of ammonia to liver
🔬 CONCEPT EXPLAINED
Ammonia is continuously produced in tissues, especially:
- Muscle
- Intestine
- Kidney
Because free ammonia is highly toxic, tissues transport it safely.
Transport via Glutamine
- Ammonia combines with glutamate
- Forms glutamine
- Glutamine travels to liver
Transport via Alanine (Glucose-Alanine Cycle)
- Muscle transfers amino group to pyruvate
- Alanine formed
- Alanine carries nitrogen to liver
Why It Exists
- Prevents toxic ammonia accumulation
- Safely transfers nitrogen to liver for urea formation
⚠️ IF DAMAGED
- Liver failure → ammonia accumulation
- Increased blood ammonia → cerebral edema
- Hyperammonemia → confusion and coma
D. Ammonia Toxicity and Brain Effects
🧠 CORE
Ammonia Toxicity
Excess ammonia damages nervous system function.
Major Brain Effects
- Cerebral edema
- Tremors
- Confusion
- Coma
🔬 CONCEPT EXPLAINED
Brain tissue is highly sensitive to ammonia.
Ammonia combines with α-ketoglutarate to form glutamate.
This causes:
- Depletion of TCA cycle intermediates
- Reduced ATP production
- Neuronal dysfunction
Glutamate further converts to glutamine inside astrocytes.
Excess glutamine causes:
- Osmotic swelling
- Cerebral edema
Why Brain Is Affected Most
- Brain depends heavily on aerobic metabolism.
- ATP depletion rapidly impairs neuronal activity.
⚠️ IF DAMAGED
Cause → Effect
- Liver failure → hyperammonemia
- Hyperammonemia → astrocyte swelling
- Cerebral edema → increased intracranial pressure
- Severe toxicity → hepatic encephalopathy and coma
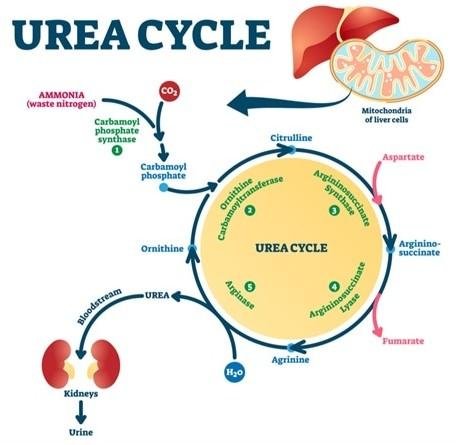
E. Urea Cycle
🧠 CORE
Definition
Metabolic pathway converting toxic ammonia into urea in liver.
Location
- Liver
- Mitochondria and cytosol
Primary Function
- Detoxification of ammonia
Major Enzymes
- Carbamoyl phosphate synthetase I
- Ornithine transcarbamylase
- Argininosuccinate synthetase
- Argininosuccinate lyase
- Arginase
🔬 CONCEPT EXPLAINED
The urea cycle is also called Krebs-Henseleit cycle.
It converts:
- Ammonia
- CO₂
into urea for renal excretion.
Steps of Urea Formation
Step 1
Ammonia + CO₂ form carbamoyl phosphate
Enzyme: Carbamoyl phosphate synthetase I
Step 2
Carbamoyl phosphate combines with ornithine
Forms citrulline
Step 3
Citrulline combines with aspartate
Forms argininosuccinate
Step 4
Argininosuccinate splits into:
- Arginine
- Fumarate
Step 5
Arginine breaks into:
- Urea
- Ornithine
Ornithine re-enters cycle.
Why It Exists
- Protects body from ammonia toxicity
- Maintains nitrogen balance
Structure → Function
- Mitochondrial steps initiate ammonia handling rapidly.
- Cytosolic steps complete urea synthesis efficiently.
⚠️ IF DAMAGED
Urea Cycle Enzyme Defects
- Hyperammonemia
- Vomiting
- Cerebral edema
- Mental retardation
- Convulsions
Important Disorder
Ornithine transcarbamylase deficiency:
- Most common inherited urea cycle disorder
- Causes severe hyperammonemia
⚙️ 4️⃣ Functional Flow
Structure → Function → Outcome
Liver
- Contains transaminases and urea cycle enzymes
→ Detoxifies ammonia
→ Maintains nitrogen balance
Muscle
- Major site of amino acid breakdown
→ Produces alanine and glutamine
→ Safe nitrogen transport
Brain
- Sensitive to ammonia
→ Requires stable ATP production
→ Hyperammonemia causes encephalopathy
Kidney
- Excretes urea
→ Removes nitrogen waste
→ Maintains metabolic homeostasis
🩺 5️⃣ Clinical Correlation
Hepatic Encephalopathy
- Liver failure causes ammonia accumulation.
- Leads to confusion, tremors, altered consciousness, coma.
Hyperammonemia
- Seen in severe liver disease and urea cycle defects.
- Causes cerebral edema and neurological symptoms.
Elevated ALT and AST
- Indicators of liver injury.
- Common in hepatitis and liver necrosis.
Urea Cycle Disorders
- Inherited enzyme deficiencies.
- Present in infancy with vomiting, lethargy, seizures.
📌 6️⃣ Summary Points